To our knowledge, there is no information about the complement system operation at pHs different from 7.4. If the complement is really dangerous to the insects in order to “force” them to produce inhibitors, complement should be active in the midgut conditions such as in pHs around 7.0 in triatomines and even in alkaline environments as in the abdominal midgut from some mosquito species where the pH reaches values equal or higher than 8.0 after a blood meal. Once the intestinal epithelium from insects has a unique cell layer, the damage caused by complement activation could lead to the GDC-0879 rupture of the digestive tract and even death of the insect. The results obtained here about the performance of the complement system at different pHs show that the classical and Doxorubicin alternative pathways are active at the pH 7.16, inside the anterior midgut from triatomines, and even at pH,8.0, inside the midgut of mosquitoes. It is possible that, under these circumstances, the alternative pathway would be triggered by carbohydrates from the glycocalix of the intestinal cells and that the classical pathway would be triggered by unspecific binding of natural antibodies to these carbohydrates or other intestinal molecules. The presence of carbohydrates covering the intestinal membranes could trigger the lectin pathway by binding the MBL protein. Figure 4 contains a scheme of the complement activation process in both, classical and alternative pathways and shows the MAC formation by the action of C3 convertases. C3 convertases also operates as C5 convertases. To the sake of simplicity, the activation of the lectin pathway was omitted. The MannamBinding Lectin pathway is not activated by any of the protocols used for the inhibitory assays in the present work. Therefore, the results obtained had no influence from this pathway. The red marks in the scheme indicate the most probable points where the salivary and/or intestinal inhibitors may be acting along the complement cascade taking into account the results observed in Tables 1 and 2. The inhibition of the C4b deposition in the classical pathway indicates a possible action over C1r and/or C1s, by inhibiting their proteolytic action as is performed by the soluble C1 inhibitor present in the normal sera. The blockage of C3b deposition in the classical pathway could be attributed to the action of the inhibitors in any point mentioned before or in the C2a proteolytic activity over C3. The presence of any factor accelerating the C2a decay from the C4bC2a-C3b complex could also lead to the same result. An inhibitor able to accelerate the decay of C2a would be similar to the C4 binding protein, which is found soluble in the normal sera. C4bp accelerates the C2a decay from the C4b-C2a-C3b complex and acts as cofactor in the cleavage of C4b by factor I, another soluble regulatory protein. The inhibition of C3b deposition in the alternative pathway could be explained by the direct inhibition of protease D. Protease D is a soluble enzyme, already active in the blood, which has higher specificity to activate factor B, by proteolysis, when it is associated to C3b opsonized to the activator surface or when it is part of the soluble complex B-C3-H2O. The activated factor B is another protease able to activate C3 to C3b and C3a. Bb inhibition could also promote reduction in C3b deposition onto activator surfaces. Any molecule, present in saliva or intestinal contents from insects, acting over the C3b-Bb-C3b complex and accelerating the decay of Bb would also favour the reduction of C3b deposition in the alternative pathway. Although the material obtained from midgut microvillosities are not able to inhibit C3b deposition by the classical pathway, we can not discard the possible presence of a MAC-assembling inhibitor inserted on the midgut membranes, as observed on erythrocytes 
Author: neuroscience research
To define the underlying mechanism of Metnase dependent adriamycin resistance
Compared to vector controls, cells with reduced NVP-BKM120 cost Metnase levels showed a 17-fold higher frequency of apoptosis after adriamycin exposure. This finding suggests that Metnase suppresses adriamycin-induced apoptosis, contributing to the increased resistance of breast cancer cells to this drug. We examined the effect of Metnase on adriamycin inhibition of Topo IIa-mediated decatention using a Temozolomide kinetoplast DNA in vitro decatenation assay. Topo IIa decatenates kDNA and adriamycin completely inhibits this activity. As shown previously, purified Metnase does not decatenate kDNA on its own, but enhances Topo IIa-dependent kDNA decatenation by 4-fold. Importantly, when Metnase is present, it overcomes the inhibition of Topo IIa by adriamycin, and this is true whether Metnase is added to the reaction before or after adriamycin. Note also that in the presence of Metnase, there is a greater level of decatentation in the presence of adriamycin than with Topo IIa alone ![]() in the absence of adriamycin. Metnase is a known component of the DSB repair pathway, and may enhance resistance to Topo IIa inhibitors by two mechanisms, enhancing DSB repair or enhancing Topo IIa function. The data presented here suggest that the ability of Metnase to interact with Topo IIa, and enhance Topo IIa dependent decatenation in vivo and in vitro may be at least as important as its ability to promote DSB repair in surviving exposure to clinical Topo IIa inhibitors. It is possible that Metnase could bind Topo IIa and physically block binding by adriamycin. In this model, Metnase would be bound to Topo IIa on DNA, and prevent adriamycin from stabilizing the Topo IIa/DNA cleavage complex, allowing Topo IIa to complete re-ligation. Alternatively, Metnase may function as a co-factor or chaperone to increase Topo IIa reaction kinetics. Here Metnase would bind transiently to Topo IIa and increase its reaction rate regardless of adriamycin binding. The mechanism may also be a functional combination of these two mechanisms where Metnase increases Topo IIa kinetics while also blocking further binding of the drug. Our interpretation of these data is that Metnase increases the intrinsic function of Topo IIa via one of the above mentioned molecular mechanisms, and that this will result in fewer DSBs, not necessarily from enhanced DNA repair, but from Topo IIa directly resisting adriamycin inhibition and thus inhibiting the production of DSBs. This model is supported by our findings that Metnase significantly blocks breast cancer cell metaphase arrest induced by ICRF-193, and that cellular resistance to Topo IIa inhibitors is directly proportional to the Metnase expression level. Our data reveal a novel mechanism for adriamycin resistance in breast cancer cells that may have important clinical implications. Metnase may be a critical biomarker for predicting tumor response to Topo IIa inhibitors. By monitoring Metnase levels, treatments with Topo IIa inhibitors may be tailored to improve efficacy. In addition, since reduced Metnase levels increase sensitivity to clinical Topo IIa inhibitors, inhibiting Metnase with a small molecule could improve response in combination therapies. Metnase inhibition may be especially important in a recurrent breast tumor that was previously exposed to Topo IIa inhibitors, since resistance to these agents may be due to upregulation of Metnase and/or Topo IIa. In summary, Metnase mediates the ability of Topo IIa to resist clinically relevant inhibitors, and may itself prove clinically useful in the treatment of breast cancer. Translationally controlled tumor protein is expressed in almost all mammalian tissues. Intracellular TCTP levels respond to various extracellular signals and agents such as growth factors, cytokines, and stress conditions. Extracellular TCTP has also been reported to be present in the supernatants of human U937 macrophage cell cultures, outside of mononuclear cells and platelets, in nasal washings, skin blister fluids, and bronchoalveolar lavage fluids during late allergic reactions.
in the absence of adriamycin. Metnase is a known component of the DSB repair pathway, and may enhance resistance to Topo IIa inhibitors by two mechanisms, enhancing DSB repair or enhancing Topo IIa function. The data presented here suggest that the ability of Metnase to interact with Topo IIa, and enhance Topo IIa dependent decatenation in vivo and in vitro may be at least as important as its ability to promote DSB repair in surviving exposure to clinical Topo IIa inhibitors. It is possible that Metnase could bind Topo IIa and physically block binding by adriamycin. In this model, Metnase would be bound to Topo IIa on DNA, and prevent adriamycin from stabilizing the Topo IIa/DNA cleavage complex, allowing Topo IIa to complete re-ligation. Alternatively, Metnase may function as a co-factor or chaperone to increase Topo IIa reaction kinetics. Here Metnase would bind transiently to Topo IIa and increase its reaction rate regardless of adriamycin binding. The mechanism may also be a functional combination of these two mechanisms where Metnase increases Topo IIa kinetics while also blocking further binding of the drug. Our interpretation of these data is that Metnase increases the intrinsic function of Topo IIa via one of the above mentioned molecular mechanisms, and that this will result in fewer DSBs, not necessarily from enhanced DNA repair, but from Topo IIa directly resisting adriamycin inhibition and thus inhibiting the production of DSBs. This model is supported by our findings that Metnase significantly blocks breast cancer cell metaphase arrest induced by ICRF-193, and that cellular resistance to Topo IIa inhibitors is directly proportional to the Metnase expression level. Our data reveal a novel mechanism for adriamycin resistance in breast cancer cells that may have important clinical implications. Metnase may be a critical biomarker for predicting tumor response to Topo IIa inhibitors. By monitoring Metnase levels, treatments with Topo IIa inhibitors may be tailored to improve efficacy. In addition, since reduced Metnase levels increase sensitivity to clinical Topo IIa inhibitors, inhibiting Metnase with a small molecule could improve response in combination therapies. Metnase inhibition may be especially important in a recurrent breast tumor that was previously exposed to Topo IIa inhibitors, since resistance to these agents may be due to upregulation of Metnase and/or Topo IIa. In summary, Metnase mediates the ability of Topo IIa to resist clinically relevant inhibitors, and may itself prove clinically useful in the treatment of breast cancer. Translationally controlled tumor protein is expressed in almost all mammalian tissues. Intracellular TCTP levels respond to various extracellular signals and agents such as growth factors, cytokines, and stress conditions. Extracellular TCTP has also been reported to be present in the supernatants of human U937 macrophage cell cultures, outside of mononuclear cells and platelets, in nasal washings, skin blister fluids, and bronchoalveolar lavage fluids during late allergic reactions.
Initiated that are designed to examine the efficacy of dietary curcumin in slowing or reversing cognitive decline
In general, curcumin studies have demonstrated that dietary administration of the compound in doses up to 12 g per day is well tolerated; however, its effects on slowing or reversing cognitive decline have been modest at best and very often dependent on the stage of AD when treatment commences. For example, in an Asian study of 1,010 non-demented individuals, a small but statistically significant improvement in cognitive abilities was noted in a population that consumed curry more than once per month. By contrast, in a more recent six-month randomized study, patients with moderateto-severe Alzheimer’s disease showed little or no measureable improvement when compared with placebo controls. These clinical findings conflict with data obtained from curcumin-treated animal models and suggest challenges lie ahead in translating findings from rodent studies to human trials. Perhaps these challenges can be met by more clearly defining the objective of curcumin treatment; either as a preventative to delay or avert the onset of significant cognitive impairment in early stage AD patients or as a therapeutic aimed at reversing the clinical hallmarks of dementia found in more advanced stages. Thus far, the majority of rodent studies have been carried out by Silmitasertib administering curcumin to animals prior to their developing AD pathologies, whereas the majority of human trials that have been attempted largely recruit individuals who are already symptomatic of AD and likely to have significant amyloid plaque burden. Reversing an already substantial plaque load may require multiple therapeutic modalities to supplement curcumin’s bioactivity or, alternatively, a more effective compound targeting Ab plaque development such as the improved inhibitor 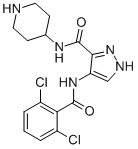 presented here. Tuberculosis is still a worldwide problem as the number of new cases continues to grow, approaching 9.8 million in 2010 and resulting in approximately 1.68 million deaths in 2009. Human immunodeficiency virus co-infection is a crucial factor in the rise in the number of TB cases and the development of active tuberculosis. In addition, multidrug resistant and extensively drug resistant strains continue to evolve, making current treatments ineffective. To counter the drug resistance problem there is a crucial need to identify new drug targets. Inosine monophosphate is obtained in mycobacteria by the de novo purine nucleotide biosynthesis pathway wherein the purine ring is assembled in a stepwise manner starting from phosphoribosyl pyrophosphate through eleven distinct enzymatic steps. IMP is a common precursor for both adenine and guanine nucleotide synthesis. The first of the two steps towards guanine nucleotide biosynthesis is CHIR-99021 252917-06-9 catalysed by inosine monophosphate dehydrogenase which converts IMP to xanthosine monophosphate with the concomitant conversion of NAD+ to NADH. The IMPDH reaction equilibrium strongly favors the forward reaction and maintains the guanine nucleotide pool. In M. tuberculosis Mt-GuaB2 is solely responsible for this essential function, since out of the three genes that encode IMPDH Mt-GuaB2 is the only functional ortholog. IMPDH is considered an attractive target for immunosuppressive, cancer, antiviral, and antimicrobial therapy. A genome wide transposon mutagenesis study indicated that M. tuberculosis requires Mt-GuaB2 for its survival. IMPDH inhibitors cause a reduction of guanine nucleotide levels and increase adenine nucleotides in vivo, and subsequently, DNA and RNA synthesis is interrupted resulting in cytotoxicity. Depending on the mode of enzyme binding, IMPDH inhibitors are classified into three types: type I inhibitors are IMP/XMP analogues, type II are NADH analogues and type III are multisubstrate inhibitors.
presented here. Tuberculosis is still a worldwide problem as the number of new cases continues to grow, approaching 9.8 million in 2010 and resulting in approximately 1.68 million deaths in 2009. Human immunodeficiency virus co-infection is a crucial factor in the rise in the number of TB cases and the development of active tuberculosis. In addition, multidrug resistant and extensively drug resistant strains continue to evolve, making current treatments ineffective. To counter the drug resistance problem there is a crucial need to identify new drug targets. Inosine monophosphate is obtained in mycobacteria by the de novo purine nucleotide biosynthesis pathway wherein the purine ring is assembled in a stepwise manner starting from phosphoribosyl pyrophosphate through eleven distinct enzymatic steps. IMP is a common precursor for both adenine and guanine nucleotide synthesis. The first of the two steps towards guanine nucleotide biosynthesis is CHIR-99021 252917-06-9 catalysed by inosine monophosphate dehydrogenase which converts IMP to xanthosine monophosphate with the concomitant conversion of NAD+ to NADH. The IMPDH reaction equilibrium strongly favors the forward reaction and maintains the guanine nucleotide pool. In M. tuberculosis Mt-GuaB2 is solely responsible for this essential function, since out of the three genes that encode IMPDH Mt-GuaB2 is the only functional ortholog. IMPDH is considered an attractive target for immunosuppressive, cancer, antiviral, and antimicrobial therapy. A genome wide transposon mutagenesis study indicated that M. tuberculosis requires Mt-GuaB2 for its survival. IMPDH inhibitors cause a reduction of guanine nucleotide levels and increase adenine nucleotides in vivo, and subsequently, DNA and RNA synthesis is interrupted resulting in cytotoxicity. Depending on the mode of enzyme binding, IMPDH inhibitors are classified into three types: type I inhibitors are IMP/XMP analogues, type II are NADH analogues and type III are multisubstrate inhibitors.
PDH inhibitor was the mold metabolite mycophenolic acid which is a type II inhibitor
MPA requires no metabolic activation and binds  at the NAD+ site. Other type II inhibitors like tiazofurin and selenazofurin must first be metabolically activated to adenine dinucleotides, thiazole-4-carboxamide adenine dinucleotide and selenazole-4-carboxamide adenine dinucleotide in vivo to become inhibitors. The nucleoside analogue tiazofurin and its derivatives are uncompetitive inhibitors. Typical type I inhibitors such as ribavirin and mizoribine bind at the substrate site. MPA inhibits by trapping enzyme-XMP* as a covalent intermediate, and the pattern of inhibition is uncompetitive with respect to both the substrates IMP and NAD+ due to the strong preference for E-XMP*. MPA and mizoribine are used in immunosuppressive chemotherapy and ribavirin for antiviral chemotherapy. Mizoribine, an IMP analogue, is a potent Kinase Inhibitor Library inhibitor of microbial enzymes. The phenyloxazole urea scaffolds were discovered in a structure-based drug design effort at Vertex Pharmaceuticals. Like MPA, these compounds trap the covalent intermediate E-XMP* complex. Imidazodiazapine nucleotide is a potent inhibitor of Escherichia coli IMPDH. Although halicyclamine was originally identified as a human IMPDH type II inhibitor, it was recently found that the antitubercular activity of halicyclamine was not due to inhibition of IMPDH. The first potent inhibitors of MtGuaB2 reported were the triazole linked mycophenolic adenine dinucleotides which showed uncompetitive inhibition with both NAD + and IMP. Recently, several analogues in the diphenyl urea class of Mt-GuaB2 inhibitors were selected based on their potent antitubercular activity and informatics analysis. Among the characterized bacterial IMPDH enzymes are those from E. coli, Streptococcus pyogenes, Streptococcus suis, Bacillus subtilis, Borrelia burgdorferi, Halobacterium salinarum and M. tuberculosis. IMPDH exists as a homotetramer. Each monomer consists of two domains: the larger catalytic core domain which forms an 8 barrel and a smaller subdomain containing two cystathionine b synthase domains also called the bateman domain. The subdomain is not required for activity although still present in all the IMPDHs characterized to date. In E. coli the subdomain is known to regulate the distribution of adenine and guanine nucleotide pools. The larger domain contains an active site loop at the C-terminal end of the b barrel strands. The substrates, IMP and NAD+ bind to the active site and, following NADH release, E-XMP* is hydrolysed. During the enzymatic oxidation of IMP to XMP, the active site cysteine residue is covalently modified. In order to preselect for Mt-GuaB2 inhibitors that show antibacterial activity, we selected scaffolds based on whole cell antibacterial data from our previous M. tuberculosis H37Rv screens of three libraries: the NIH Molecular CPI-613 95809-78-2 libraries Small Molecule Repository, the Life Chemicals kinase library and an in house Chembridge library. All compound selections were made from active compounds and full dose-response data from these screens: 2273 actives identified from the MLSMR, 1781 from the Chembridge set and 1329 from the kinase library. Only a small number of nonnucleoside, small molecule IMPDH ligands has been published for various species. We utilized core scaffolds of these known IMPDH ligands for searching our TB active sets for potential M. tuberculosis IMPDH inhibitors. The search resulted in the identification of five analogues of the known IMPDH inhibitor scaffold 2-phenoxy-N-phenylpropanamide and these compounds were included in the set of compounds evaluated in this study. A focused scaffold-based approach was applied to select further compounds that also possess structural novelty as potential MtGuaB2 inhibitors.
at the NAD+ site. Other type II inhibitors like tiazofurin and selenazofurin must first be metabolically activated to adenine dinucleotides, thiazole-4-carboxamide adenine dinucleotide and selenazole-4-carboxamide adenine dinucleotide in vivo to become inhibitors. The nucleoside analogue tiazofurin and its derivatives are uncompetitive inhibitors. Typical type I inhibitors such as ribavirin and mizoribine bind at the substrate site. MPA inhibits by trapping enzyme-XMP* as a covalent intermediate, and the pattern of inhibition is uncompetitive with respect to both the substrates IMP and NAD+ due to the strong preference for E-XMP*. MPA and mizoribine are used in immunosuppressive chemotherapy and ribavirin for antiviral chemotherapy. Mizoribine, an IMP analogue, is a potent Kinase Inhibitor Library inhibitor of microbial enzymes. The phenyloxazole urea scaffolds were discovered in a structure-based drug design effort at Vertex Pharmaceuticals. Like MPA, these compounds trap the covalent intermediate E-XMP* complex. Imidazodiazapine nucleotide is a potent inhibitor of Escherichia coli IMPDH. Although halicyclamine was originally identified as a human IMPDH type II inhibitor, it was recently found that the antitubercular activity of halicyclamine was not due to inhibition of IMPDH. The first potent inhibitors of MtGuaB2 reported were the triazole linked mycophenolic adenine dinucleotides which showed uncompetitive inhibition with both NAD + and IMP. Recently, several analogues in the diphenyl urea class of Mt-GuaB2 inhibitors were selected based on their potent antitubercular activity and informatics analysis. Among the characterized bacterial IMPDH enzymes are those from E. coli, Streptococcus pyogenes, Streptococcus suis, Bacillus subtilis, Borrelia burgdorferi, Halobacterium salinarum and M. tuberculosis. IMPDH exists as a homotetramer. Each monomer consists of two domains: the larger catalytic core domain which forms an 8 barrel and a smaller subdomain containing two cystathionine b synthase domains also called the bateman domain. The subdomain is not required for activity although still present in all the IMPDHs characterized to date. In E. coli the subdomain is known to regulate the distribution of adenine and guanine nucleotide pools. The larger domain contains an active site loop at the C-terminal end of the b barrel strands. The substrates, IMP and NAD+ bind to the active site and, following NADH release, E-XMP* is hydrolysed. During the enzymatic oxidation of IMP to XMP, the active site cysteine residue is covalently modified. In order to preselect for Mt-GuaB2 inhibitors that show antibacterial activity, we selected scaffolds based on whole cell antibacterial data from our previous M. tuberculosis H37Rv screens of three libraries: the NIH Molecular CPI-613 95809-78-2 libraries Small Molecule Repository, the Life Chemicals kinase library and an in house Chembridge library. All compound selections were made from active compounds and full dose-response data from these screens: 2273 actives identified from the MLSMR, 1781 from the Chembridge set and 1329 from the kinase library. Only a small number of nonnucleoside, small molecule IMPDH ligands has been published for various species. We utilized core scaffolds of these known IMPDH ligands for searching our TB active sets for potential M. tuberculosis IMPDH inhibitors. The search resulted in the identification of five analogues of the known IMPDH inhibitor scaffold 2-phenoxy-N-phenylpropanamide and these compounds were included in the set of compounds evaluated in this study. A focused scaffold-based approach was applied to select further compounds that also possess structural novelty as potential MtGuaB2 inhibitors.
Well controlled and allows normal regulation of cellular responses as in the case of the p53 negative regulator
Increased Necdin could paradoxically promote growth or survival. A possible role for Necdin in DNA damage response was suggested by the upregulation of Necdin following different genotoxic stresses. By using nutlin-3, we showed that p53 activation clearly induced Necdin in a dose dependent manner, supporting a previous report that identified Necdin as a p53 target gene. Moreover, we show that modulation of the Necdin level affects p53-dependent growth arrest. Indeed, we demonstrate that an increase in Necdin expression results in a delayed cell cycle arrest while inversely targeting Necdin by shRNA accelerates this arrest. The interaction of Necdin with p53 suggests that this delay in growth arrest is probably associated with a direct inhibitory effect of Necdin over p53. We noted that Necdin affected p21 induction following p53 activation in our model supporting previous results. Therefore, interference with p53 transcriptional Sorafenib moa activity may represent the mechanism underlying the cell cycle arrest variations caused by Necdin. However, 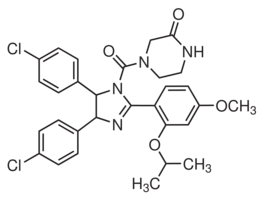 we believe that other mechanisms may be involved since p21 mediated-arrest mostly relies on functional Rb and in PyLT-expressing cells, the Rb proteins are kept inactive by their interaction with PyLT. As p53 induction upon genotoxic stress is associated with multiple additional signaling events, we directly addressed p53 stimulation by exposure to nutlin-3. This specific stimulation results in a functional induction of p53, although the posttranslational SCH727965 CDK inhibitor phosphorylation of p53 observed with genotoxic stress are absent or barely detectable with nutlin-3. This suggests that phosphorylation may not be critical for interaction of Necdin with p53 and that Necdin does not interfere with the phosphorylation status to modulate p53 activity. In addition to phosphorylation, other modifications contribute to p53 activity, including acetylation, which is increased upon nutlin-3 stimulation. The deacetylase Sirt1 is a negative regulator of p53 activation and Necdin interactions with this protein potentiate its activity upon genotoxic stress. However, we did not address the status of these post-translationals modifications in our model. Additionally, it is important to note that p53 responses can differ with particular drugs depending on the dose employed, the duration of the treatment, and the metabolic state of the cell. Others mechanisms can explain Necdin inhibitory effects over p53. Necdin binds the N-terminal transactivation domain of p53. Some proteins share this binding site, among them SOCS1, which contribute to p53 activation. It is possible that Necdin competes with activating proteins such as SOCS1 for p53 binding, leading to a decrease in p53 response. Others examples exist where the response to p53 activation varies according to the presence or absence of specific cellular partners. The capacity of p53 to translocate to the mitochondria where it plays a transcription-independent function in apoptosis is now well documented. Tid1 is a p53-interacting protein that helps this localization from the nucleus to mitochondria. Both cytoplasmic and nuclear cellular partners have been revealed for Necdin and expression of these partners has been shown to cause Necdin relocalisation in the cell. Perhaps interference with p53 activation may arise from the ability of Necdin to relocate p53 in other cellular compartment. All these mechanisms are consistent with the notion that Necdin can inhibit p53 function and require further investigation. Combining our data on p53 inhibition by Necdin with the knowledge that Necdin is a direct p53 response gene suggests that Necdin is part of a negative feedback loop controlling p53 activity.
we believe that other mechanisms may be involved since p21 mediated-arrest mostly relies on functional Rb and in PyLT-expressing cells, the Rb proteins are kept inactive by their interaction with PyLT. As p53 induction upon genotoxic stress is associated with multiple additional signaling events, we directly addressed p53 stimulation by exposure to nutlin-3. This specific stimulation results in a functional induction of p53, although the posttranslational SCH727965 CDK inhibitor phosphorylation of p53 observed with genotoxic stress are absent or barely detectable with nutlin-3. This suggests that phosphorylation may not be critical for interaction of Necdin with p53 and that Necdin does not interfere with the phosphorylation status to modulate p53 activity. In addition to phosphorylation, other modifications contribute to p53 activity, including acetylation, which is increased upon nutlin-3 stimulation. The deacetylase Sirt1 is a negative regulator of p53 activation and Necdin interactions with this protein potentiate its activity upon genotoxic stress. However, we did not address the status of these post-translationals modifications in our model. Additionally, it is important to note that p53 responses can differ with particular drugs depending on the dose employed, the duration of the treatment, and the metabolic state of the cell. Others mechanisms can explain Necdin inhibitory effects over p53. Necdin binds the N-terminal transactivation domain of p53. Some proteins share this binding site, among them SOCS1, which contribute to p53 activation. It is possible that Necdin competes with activating proteins such as SOCS1 for p53 binding, leading to a decrease in p53 response. Others examples exist where the response to p53 activation varies according to the presence or absence of specific cellular partners. The capacity of p53 to translocate to the mitochondria where it plays a transcription-independent function in apoptosis is now well documented. Tid1 is a p53-interacting protein that helps this localization from the nucleus to mitochondria. Both cytoplasmic and nuclear cellular partners have been revealed for Necdin and expression of these partners has been shown to cause Necdin relocalisation in the cell. Perhaps interference with p53 activation may arise from the ability of Necdin to relocate p53 in other cellular compartment. All these mechanisms are consistent with the notion that Necdin can inhibit p53 function and require further investigation. Combining our data on p53 inhibition by Necdin with the knowledge that Necdin is a direct p53 response gene suggests that Necdin is part of a negative feedback loop controlling p53 activity.