Several studies have uncovered physiologically important and disease relevant functions for the classic receptor type PTP, PTPs, which underscore its potential as a biological target. PTPs is highly AZ 960 expressed in neuronal tissue where it regulates axon guidance and neurite outgrowth. Furthermore, it was recently reported that loss of PTPs facilitates nerve regeneration following spinal cord injury, owing to the interaction of its ectodomain with chondroitin sulfate proteoglycans. In addition to its neural function, PTPs has been implicated in chemoresistance of cancer cells. First, we discovered that RNAi-mediated knockdown of PTPs in cultured cancer cells confers resistance to several chemotherapeutics. Additionally, we have discovered that loss of PTPs hyperactivates autophagy, a cellular recycling program that may contribute to chemoresistance of cancer cells. Taken together, it is apparent that modulation of PTPs may have therapeutic potential in a range of contexts. Notably, inhibition of PTPs could potentially provide benefit following SCI through enhanced neural regeneration. In addition, it is possible that PTPs inhibition may yield therapeutic value in diseases in which increasing autophagy represents a promising treatment strategy. Furthermore, a small Nutlin-3 moa molecule would provide value as a molecular probe or tool compound to interrogate the cellular functions and disease implications of PTPs. Several approaches exist for the identification of small molecule inhibitors of phosphatases. While high-throughput screening of compounds in vitro has been successfully utilized to discover inhibitors of LAR, PTP1B, SHP2, CD45, and others, the technical and physical investment is considerable as is the potential for experimental artifacts leading to false negatives and positives. Alternatively, a primary screen for inhibitor scaffolds can be guided by in silico virtual screening. This method involves high-throughput computational docking of small molecules into the crystal structure of a phosphatase active site and selecting the molecules which bind favorably, akin to a natural substrate. Following the selection of the best-scoring scaffolds, each scaffold can then be tested and validated for phosphatase inhibition in vitro. This approach has gained popularity as the number of enzymes with solved crystal structures has increased and it is advantageous in many ways. First, utilization of 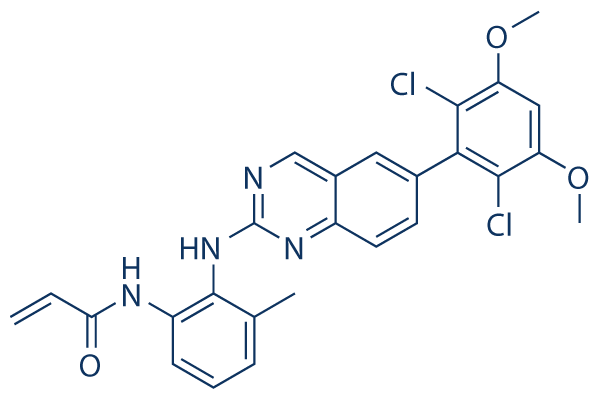 the phosphatase structure allows for the exclusion of molecules which have little chance of interacting with the active site, greatly reducing the number of scaffolds to be biochemically screened and improving the screen results. Second, an understanding of the unique structural features and residues comprising the active site as well as proximal folds or binding pockets can guide the selection and refinement of an inhibitor. Furthermore, an in silico approach is incredibly efficient in that it allows tens of thousands to millions of compounds to be screened virtually in a matter of weeks. The increasing number of PTP experimental structures resolved by X-ray crystallography has stimulated structure-guided efforts to identify small molecule PTP inhibitors. Drug discovery efforts focusing on PTPs are outlined in a comprehensive review written by Blaskovich, including detailed descriptions of the biological roles, target validation, screening tools and artifacts, and medicinal chemistry efforts, surrounding PTPs.
the phosphatase structure allows for the exclusion of molecules which have little chance of interacting with the active site, greatly reducing the number of scaffolds to be biochemically screened and improving the screen results. Second, an understanding of the unique structural features and residues comprising the active site as well as proximal folds or binding pockets can guide the selection and refinement of an inhibitor. Furthermore, an in silico approach is incredibly efficient in that it allows tens of thousands to millions of compounds to be screened virtually in a matter of weeks. The increasing number of PTP experimental structures resolved by X-ray crystallography has stimulated structure-guided efforts to identify small molecule PTP inhibitors. Drug discovery efforts focusing on PTPs are outlined in a comprehensive review written by Blaskovich, including detailed descriptions of the biological roles, target validation, screening tools and artifacts, and medicinal chemistry efforts, surrounding PTPs.
Month: August 2019
Molecular modeling efforts have primarily focused on hit generation and structure-guided optimization of hits for PTP1B
A more recent study by Park and coworkers used structure-based virtual screening to identify nine PTP1B inhibitors with significant potency. Utilizing the growing knowledge base from known PTP1B inhibitors, Suresh et al. reported the generation of a chemical feature-based pharmacophore hypothesis and its use for the identification of new lead compounds. Additional PTPs were also approached using in silico methodologies. Of particular interest was the study by Hu et al., which targeted the identification of small molecule inhibitors for bacterial Yersinia YopH and Salmonella SptP through differentiation with PTP1B. Virtual screening also identified small molecule inhibitors of LMWPTP, SHP-2, and Cdc25. A review by He and coworkers underscores the progress made to date in identifying small molecule tools for the functional interrogation of various PTPs, assisted by the computational tools. In addition to the classes listed above, in silico screening also supported the identification of Lyp inhibitors, as described in three studies by Yu, Wu, and Stanford. Importantly, the review by He articulates both the challenges and opportunities for developing PTP specific inhibitors, serving as chemical probes to augment the knowledge of PTP biology, and to establish the basis GDC-0879 needed to approach other PTPs currently underexplored. In this study, we identified small molecule inhibitors targeting the active site of PTPs. We screened compounds in silico to identify structurally distinct scaffolds predicted to have the most desirable binding energies. These PTPs virtual hits, as well as additional compounds identified by a substructure and similarity search, were iteratively tested for inhibition of PTPs in vitro. While we discovered 25 active compounds with micromolar potency against PTPs, we discovered compounds frequently catalyzed the production of oxidative species in the assay buffer, a common culprit for non-selective PTP inhibition. By optimizing the biochemical screen to include oxidation constraints, we identified one lead compound which inhibited PTPs by a mechanism that was oxidation-independent. This lead hit was capable of docking into the active site, suggesting it functions as a competitive inhibitor. The results of this study will be used as the foundation of future structure-based refinement of PTPs inhibitors. The tandem phosphatase domains of PTPs have been crystallized in their apo form. We retrieved this structure from the protein data bank and verified its utility by molecularly docking a phosphotyrosine peptide into the catalytically active D1 domain. We hypothesized that the active site could be exploited in the development of competitive inhibitors targeted to PTPs. To this end, we used the ZINC database to virtually screen a library of compounds for their ability to dock into the D1 domain of PTPs. From the top scoring compounds which were most favorably bound by the active site, we identified three compounds which represented structurally distinct scaffolds and demonstrated an ability to inhibit PTPs activity in preliminary in vitro ICG-001 cost assays. To expand these into a  set of compounds for biochemical investigation, we performed a substructure search and retrieved 74 additional molecules similar to these three scaffolds from the ChemBridge compound library.
set of compounds for biochemical investigation, we performed a substructure search and retrieved 74 additional molecules similar to these three scaffolds from the ChemBridge compound library.
Difference in tumor pO2 in rapamycin treated group to the day matched control compared to the greater
Difference in tumor pO2 in sunitinib treated group to the control. The significant synergy with mTOR inhibitors including rapamycin and radiation reported by Shinohara et al may point out the Oligomycin A 579-13-5 characteristic influences of the microenvironment of each tumor type as pointed out in other studies where the synergy was attributed only to rapamycin targeting the enhanced activity of signaling pathways controlled by mTOR in the host endothelial cells. Recent studies with a dual inhibitor of the PI3K and mTOR pathway found that the period of vascular remodeling is relatively more sustained than that observed with anti-angiogenic drugs resulting in substantial therapeutic gain. These studies point to the importance of longitudinally monitoring such changes to realize maximal efficacy in combined chemo-radiation treatments. Imaging studies of the tumor microenvironment can establish a strategy in preclinical models to identify an optimal treatment schedule to realize enhanced response to combination treatments. In summary, results from the current study show that molecular imaging techniques provide an opportunity to serially monitor changes in tumor physiology non-invasively and quantitatively and identify subtle physiological changes in response to rapamycin treatment. Therefore these techniques have the ability to provide valuable non-invasive biomarkers which predict treatment outcome and also identify temporal windows where radiation therapy can be advantageously combined to elicit improved response. Tyrosine phosphorylation is a critical mechanism by which cells exert control over signaling processes. Protein tyrosine kinases and phosphatases work in concert to control these signaling cascades, and alterations in the expression or activity of these enzymes hallmark many human diseases. While PTKs have long been the focus of extensive research and drug development efforts, the role of PTPs as critical mediators of signal transduction was initially underappreciated. Consequently, the molecular characterization of these phosphatases has trailed that of PTKs, and only recently has the PTP field reached the forefront of disease based-research. As validation for phosphastases in human disease, half of PTP genes are now implicated in at least one human disease. The critical role of PTPs in cell  function and their role in disease etiology highlight the importance of developing phosphatase agonists and inhibitors. Unfortunately, phosphatases have historically been perceived as “undruggable” for several important reasons. First, phosphatases often control multiple signaling pathways and thus, inhibition of a single enzyme may not yield a specific cellular effect. Second, signaling cascades are generally controlled by multiple phosphatases and accordingly, blocking the activity of one may not sufficiently induce the desired modulation to a signaling pathway. Finally, and most importantly, phosphatase active sites display high conservation which hinders the ability to develop catalysis-directed inhibitors with any degree of selectivity. Despite these pitfalls, the emerging role of PTPs in human disease etiology has Screening Libraries necessitated a solution. Largely through use of structure-based drug design, several PTPs now represent promising targets for disease treatment. Most notably, bidentate inhibitors of PTP1B, implicated in type II diabetes and obesity, have been developed which span both the catalytic pocket and a second substrate binding pocket discovered.
function and their role in disease etiology highlight the importance of developing phosphatase agonists and inhibitors. Unfortunately, phosphatases have historically been perceived as “undruggable” for several important reasons. First, phosphatases often control multiple signaling pathways and thus, inhibition of a single enzyme may not yield a specific cellular effect. Second, signaling cascades are generally controlled by multiple phosphatases and accordingly, blocking the activity of one may not sufficiently induce the desired modulation to a signaling pathway. Finally, and most importantly, phosphatase active sites display high conservation which hinders the ability to develop catalysis-directed inhibitors with any degree of selectivity. Despite these pitfalls, the emerging role of PTPs in human disease etiology has Screening Libraries necessitated a solution. Largely through use of structure-based drug design, several PTPs now represent promising targets for disease treatment. Most notably, bidentate inhibitors of PTP1B, implicated in type II diabetes and obesity, have been developed which span both the catalytic pocket and a second substrate binding pocket discovered.