In general, curcumin studies have demonstrated that dietary administration of the compound in doses up to 12 g per day is well tolerated; however, its effects on slowing or reversing cognitive decline have been modest at best and very often dependent on the stage of AD when treatment commences. For example, in an Asian study of 1,010 non-demented individuals, a small but statistically significant improvement in cognitive abilities was noted in a population that consumed curry more than once per month. By contrast, in a more recent six-month randomized study, patients with moderateto-severe Alzheimer’s disease showed little or no measureable improvement when compared with placebo controls. These clinical findings conflict with data obtained from curcumin-treated animal models and suggest challenges lie ahead in translating findings from rodent studies to human trials. Perhaps these challenges can be met by more clearly defining the objective of curcumin treatment; either as a preventative to delay or avert the onset of significant cognitive impairment in early stage AD patients or as a therapeutic aimed at reversing the clinical hallmarks of dementia found in more advanced stages. Thus far, the majority of rodent studies have been carried out by Silmitasertib administering curcumin to animals prior to their developing AD pathologies, whereas the majority of human trials that have been attempted largely recruit individuals who are already symptomatic of AD and likely to have significant amyloid plaque burden. Reversing an already substantial plaque load may require multiple therapeutic modalities to supplement curcumin’s bioactivity or, alternatively, a more effective compound targeting Ab plaque development such as the improved inhibitor 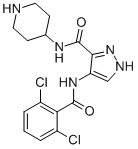 presented here. Tuberculosis is still a worldwide problem as the number of new cases continues to grow, approaching 9.8 million in 2010 and resulting in approximately 1.68 million deaths in 2009. Human immunodeficiency virus co-infection is a crucial factor in the rise in the number of TB cases and the development of active tuberculosis. In addition, multidrug resistant and extensively drug resistant strains continue to evolve, making current treatments ineffective. To counter the drug resistance problem there is a crucial need to identify new drug targets. Inosine monophosphate is obtained in mycobacteria by the de novo purine nucleotide biosynthesis pathway wherein the purine ring is assembled in a stepwise manner starting from phosphoribosyl pyrophosphate through eleven distinct enzymatic steps. IMP is a common precursor for both adenine and guanine nucleotide synthesis. The first of the two steps towards guanine nucleotide biosynthesis is CHIR-99021 252917-06-9 catalysed by inosine monophosphate dehydrogenase which converts IMP to xanthosine monophosphate with the concomitant conversion of NAD+ to NADH. The IMPDH reaction equilibrium strongly favors the forward reaction and maintains the guanine nucleotide pool. In M. tuberculosis Mt-GuaB2 is solely responsible for this essential function, since out of the three genes that encode IMPDH Mt-GuaB2 is the only functional ortholog. IMPDH is considered an attractive target for immunosuppressive, cancer, antiviral, and antimicrobial therapy. A genome wide transposon mutagenesis study indicated that M. tuberculosis requires Mt-GuaB2 for its survival. IMPDH inhibitors cause a reduction of guanine nucleotide levels and increase adenine nucleotides in vivo, and subsequently, DNA and RNA synthesis is interrupted resulting in cytotoxicity. Depending on the mode of enzyme binding, IMPDH inhibitors are classified into three types: type I inhibitors are IMP/XMP analogues, type II are NADH analogues and type III are multisubstrate inhibitors.
presented here. Tuberculosis is still a worldwide problem as the number of new cases continues to grow, approaching 9.8 million in 2010 and resulting in approximately 1.68 million deaths in 2009. Human immunodeficiency virus co-infection is a crucial factor in the rise in the number of TB cases and the development of active tuberculosis. In addition, multidrug resistant and extensively drug resistant strains continue to evolve, making current treatments ineffective. To counter the drug resistance problem there is a crucial need to identify new drug targets. Inosine monophosphate is obtained in mycobacteria by the de novo purine nucleotide biosynthesis pathway wherein the purine ring is assembled in a stepwise manner starting from phosphoribosyl pyrophosphate through eleven distinct enzymatic steps. IMP is a common precursor for both adenine and guanine nucleotide synthesis. The first of the two steps towards guanine nucleotide biosynthesis is CHIR-99021 252917-06-9 catalysed by inosine monophosphate dehydrogenase which converts IMP to xanthosine monophosphate with the concomitant conversion of NAD+ to NADH. The IMPDH reaction equilibrium strongly favors the forward reaction and maintains the guanine nucleotide pool. In M. tuberculosis Mt-GuaB2 is solely responsible for this essential function, since out of the three genes that encode IMPDH Mt-GuaB2 is the only functional ortholog. IMPDH is considered an attractive target for immunosuppressive, cancer, antiviral, and antimicrobial therapy. A genome wide transposon mutagenesis study indicated that M. tuberculosis requires Mt-GuaB2 for its survival. IMPDH inhibitors cause a reduction of guanine nucleotide levels and increase adenine nucleotides in vivo, and subsequently, DNA and RNA synthesis is interrupted resulting in cytotoxicity. Depending on the mode of enzyme binding, IMPDH inhibitors are classified into three types: type I inhibitors are IMP/XMP analogues, type II are NADH analogues and type III are multisubstrate inhibitors.
Month: August 2019
PDH inhibitor was the mold metabolite mycophenolic acid which is a type II inhibitor
MPA requires no metabolic activation and binds  at the NAD+ site. Other type II inhibitors like tiazofurin and selenazofurin must first be metabolically activated to adenine dinucleotides, thiazole-4-carboxamide adenine dinucleotide and selenazole-4-carboxamide adenine dinucleotide in vivo to become inhibitors. The nucleoside analogue tiazofurin and its derivatives are uncompetitive inhibitors. Typical type I inhibitors such as ribavirin and mizoribine bind at the substrate site. MPA inhibits by trapping enzyme-XMP* as a covalent intermediate, and the pattern of inhibition is uncompetitive with respect to both the substrates IMP and NAD+ due to the strong preference for E-XMP*. MPA and mizoribine are used in immunosuppressive chemotherapy and ribavirin for antiviral chemotherapy. Mizoribine, an IMP analogue, is a potent Kinase Inhibitor Library inhibitor of microbial enzymes. The phenyloxazole urea scaffolds were discovered in a structure-based drug design effort at Vertex Pharmaceuticals. Like MPA, these compounds trap the covalent intermediate E-XMP* complex. Imidazodiazapine nucleotide is a potent inhibitor of Escherichia coli IMPDH. Although halicyclamine was originally identified as a human IMPDH type II inhibitor, it was recently found that the antitubercular activity of halicyclamine was not due to inhibition of IMPDH. The first potent inhibitors of MtGuaB2 reported were the triazole linked mycophenolic adenine dinucleotides which showed uncompetitive inhibition with both NAD + and IMP. Recently, several analogues in the diphenyl urea class of Mt-GuaB2 inhibitors were selected based on their potent antitubercular activity and informatics analysis. Among the characterized bacterial IMPDH enzymes are those from E. coli, Streptococcus pyogenes, Streptococcus suis, Bacillus subtilis, Borrelia burgdorferi, Halobacterium salinarum and M. tuberculosis. IMPDH exists as a homotetramer. Each monomer consists of two domains: the larger catalytic core domain which forms an 8 barrel and a smaller subdomain containing two cystathionine b synthase domains also called the bateman domain. The subdomain is not required for activity although still present in all the IMPDHs characterized to date. In E. coli the subdomain is known to regulate the distribution of adenine and guanine nucleotide pools. The larger domain contains an active site loop at the C-terminal end of the b barrel strands. The substrates, IMP and NAD+ bind to the active site and, following NADH release, E-XMP* is hydrolysed. During the enzymatic oxidation of IMP to XMP, the active site cysteine residue is covalently modified. In order to preselect for Mt-GuaB2 inhibitors that show antibacterial activity, we selected scaffolds based on whole cell antibacterial data from our previous M. tuberculosis H37Rv screens of three libraries: the NIH Molecular CPI-613 95809-78-2 libraries Small Molecule Repository, the Life Chemicals kinase library and an in house Chembridge library. All compound selections were made from active compounds and full dose-response data from these screens: 2273 actives identified from the MLSMR, 1781 from the Chembridge set and 1329 from the kinase library. Only a small number of nonnucleoside, small molecule IMPDH ligands has been published for various species. We utilized core scaffolds of these known IMPDH ligands for searching our TB active sets for potential M. tuberculosis IMPDH inhibitors. The search resulted in the identification of five analogues of the known IMPDH inhibitor scaffold 2-phenoxy-N-phenylpropanamide and these compounds were included in the set of compounds evaluated in this study. A focused scaffold-based approach was applied to select further compounds that also possess structural novelty as potential MtGuaB2 inhibitors.
at the NAD+ site. Other type II inhibitors like tiazofurin and selenazofurin must first be metabolically activated to adenine dinucleotides, thiazole-4-carboxamide adenine dinucleotide and selenazole-4-carboxamide adenine dinucleotide in vivo to become inhibitors. The nucleoside analogue tiazofurin and its derivatives are uncompetitive inhibitors. Typical type I inhibitors such as ribavirin and mizoribine bind at the substrate site. MPA inhibits by trapping enzyme-XMP* as a covalent intermediate, and the pattern of inhibition is uncompetitive with respect to both the substrates IMP and NAD+ due to the strong preference for E-XMP*. MPA and mizoribine are used in immunosuppressive chemotherapy and ribavirin for antiviral chemotherapy. Mizoribine, an IMP analogue, is a potent Kinase Inhibitor Library inhibitor of microbial enzymes. The phenyloxazole urea scaffolds were discovered in a structure-based drug design effort at Vertex Pharmaceuticals. Like MPA, these compounds trap the covalent intermediate E-XMP* complex. Imidazodiazapine nucleotide is a potent inhibitor of Escherichia coli IMPDH. Although halicyclamine was originally identified as a human IMPDH type II inhibitor, it was recently found that the antitubercular activity of halicyclamine was not due to inhibition of IMPDH. The first potent inhibitors of MtGuaB2 reported were the triazole linked mycophenolic adenine dinucleotides which showed uncompetitive inhibition with both NAD + and IMP. Recently, several analogues in the diphenyl urea class of Mt-GuaB2 inhibitors were selected based on their potent antitubercular activity and informatics analysis. Among the characterized bacterial IMPDH enzymes are those from E. coli, Streptococcus pyogenes, Streptococcus suis, Bacillus subtilis, Borrelia burgdorferi, Halobacterium salinarum and M. tuberculosis. IMPDH exists as a homotetramer. Each monomer consists of two domains: the larger catalytic core domain which forms an 8 barrel and a smaller subdomain containing two cystathionine b synthase domains also called the bateman domain. The subdomain is not required for activity although still present in all the IMPDHs characterized to date. In E. coli the subdomain is known to regulate the distribution of adenine and guanine nucleotide pools. The larger domain contains an active site loop at the C-terminal end of the b barrel strands. The substrates, IMP and NAD+ bind to the active site and, following NADH release, E-XMP* is hydrolysed. During the enzymatic oxidation of IMP to XMP, the active site cysteine residue is covalently modified. In order to preselect for Mt-GuaB2 inhibitors that show antibacterial activity, we selected scaffolds based on whole cell antibacterial data from our previous M. tuberculosis H37Rv screens of three libraries: the NIH Molecular CPI-613 95809-78-2 libraries Small Molecule Repository, the Life Chemicals kinase library and an in house Chembridge library. All compound selections were made from active compounds and full dose-response data from these screens: 2273 actives identified from the MLSMR, 1781 from the Chembridge set and 1329 from the kinase library. Only a small number of nonnucleoside, small molecule IMPDH ligands has been published for various species. We utilized core scaffolds of these known IMPDH ligands for searching our TB active sets for potential M. tuberculosis IMPDH inhibitors. The search resulted in the identification of five analogues of the known IMPDH inhibitor scaffold 2-phenoxy-N-phenylpropanamide and these compounds were included in the set of compounds evaluated in this study. A focused scaffold-based approach was applied to select further compounds that also possess structural novelty as potential MtGuaB2 inhibitors.
Well controlled and allows normal regulation of cellular responses as in the case of the p53 negative regulator
Increased Necdin could paradoxically promote growth or survival. A possible role for Necdin in DNA damage response was suggested by the upregulation of Necdin following different genotoxic stresses. By using nutlin-3, we showed that p53 activation clearly induced Necdin in a dose dependent manner, supporting a previous report that identified Necdin as a p53 target gene. Moreover, we show that modulation of the Necdin level affects p53-dependent growth arrest. Indeed, we demonstrate that an increase in Necdin expression results in a delayed cell cycle arrest while inversely targeting Necdin by shRNA accelerates this arrest. The interaction of Necdin with p53 suggests that this delay in growth arrest is probably associated with a direct inhibitory effect of Necdin over p53. We noted that Necdin affected p21 induction following p53 activation in our model supporting previous results. Therefore, interference with p53 transcriptional Sorafenib moa activity may represent the mechanism underlying the cell cycle arrest variations caused by Necdin. However,  we believe that other mechanisms may be involved since p21 mediated-arrest mostly relies on functional Rb and in PyLT-expressing cells, the Rb proteins are kept inactive by their interaction with PyLT. As p53 induction upon genotoxic stress is associated with multiple additional signaling events, we directly addressed p53 stimulation by exposure to nutlin-3. This specific stimulation results in a functional induction of p53, although the posttranslational SCH727965 CDK inhibitor phosphorylation of p53 observed with genotoxic stress are absent or barely detectable with nutlin-3. This suggests that phosphorylation may not be critical for interaction of Necdin with p53 and that Necdin does not interfere with the phosphorylation status to modulate p53 activity. In addition to phosphorylation, other modifications contribute to p53 activity, including acetylation, which is increased upon nutlin-3 stimulation. The deacetylase Sirt1 is a negative regulator of p53 activation and Necdin interactions with this protein potentiate its activity upon genotoxic stress. However, we did not address the status of these post-translationals modifications in our model. Additionally, it is important to note that p53 responses can differ with particular drugs depending on the dose employed, the duration of the treatment, and the metabolic state of the cell. Others mechanisms can explain Necdin inhibitory effects over p53. Necdin binds the N-terminal transactivation domain of p53. Some proteins share this binding site, among them SOCS1, which contribute to p53 activation. It is possible that Necdin competes with activating proteins such as SOCS1 for p53 binding, leading to a decrease in p53 response. Others examples exist where the response to p53 activation varies according to the presence or absence of specific cellular partners. The capacity of p53 to translocate to the mitochondria where it plays a transcription-independent function in apoptosis is now well documented. Tid1 is a p53-interacting protein that helps this localization from the nucleus to mitochondria. Both cytoplasmic and nuclear cellular partners have been revealed for Necdin and expression of these partners has been shown to cause Necdin relocalisation in the cell. Perhaps interference with p53 activation may arise from the ability of Necdin to relocate p53 in other cellular compartment. All these mechanisms are consistent with the notion that Necdin can inhibit p53 function and require further investigation. Combining our data on p53 inhibition by Necdin with the knowledge that Necdin is a direct p53 response gene suggests that Necdin is part of a negative feedback loop controlling p53 activity.
we believe that other mechanisms may be involved since p21 mediated-arrest mostly relies on functional Rb and in PyLT-expressing cells, the Rb proteins are kept inactive by their interaction with PyLT. As p53 induction upon genotoxic stress is associated with multiple additional signaling events, we directly addressed p53 stimulation by exposure to nutlin-3. This specific stimulation results in a functional induction of p53, although the posttranslational SCH727965 CDK inhibitor phosphorylation of p53 observed with genotoxic stress are absent or barely detectable with nutlin-3. This suggests that phosphorylation may not be critical for interaction of Necdin with p53 and that Necdin does not interfere with the phosphorylation status to modulate p53 activity. In addition to phosphorylation, other modifications contribute to p53 activity, including acetylation, which is increased upon nutlin-3 stimulation. The deacetylase Sirt1 is a negative regulator of p53 activation and Necdin interactions with this protein potentiate its activity upon genotoxic stress. However, we did not address the status of these post-translationals modifications in our model. Additionally, it is important to note that p53 responses can differ with particular drugs depending on the dose employed, the duration of the treatment, and the metabolic state of the cell. Others mechanisms can explain Necdin inhibitory effects over p53. Necdin binds the N-terminal transactivation domain of p53. Some proteins share this binding site, among them SOCS1, which contribute to p53 activation. It is possible that Necdin competes with activating proteins such as SOCS1 for p53 binding, leading to a decrease in p53 response. Others examples exist where the response to p53 activation varies according to the presence or absence of specific cellular partners. The capacity of p53 to translocate to the mitochondria where it plays a transcription-independent function in apoptosis is now well documented. Tid1 is a p53-interacting protein that helps this localization from the nucleus to mitochondria. Both cytoplasmic and nuclear cellular partners have been revealed for Necdin and expression of these partners has been shown to cause Necdin relocalisation in the cell. Perhaps interference with p53 activation may arise from the ability of Necdin to relocate p53 in other cellular compartment. All these mechanisms are consistent with the notion that Necdin can inhibit p53 function and require further investigation. Combining our data on p53 inhibition by Necdin with the knowledge that Necdin is a direct p53 response gene suggests that Necdin is part of a negative feedback loop controlling p53 activity.
Thereby either our model or the crystal structure should be considered equally representative
The work presented here supports core, the HCV capsid protein as a novel target for anti-HCV drug development. We show that an inhibitor of capsid protein dimerization can specifically and directly bind to core and core-based complexes with other HCV proteins. This binding possibly results in disruption of assembly or in disassembly of the viral particle, leading to reduction of infective HCV particles. One added advantage of HCV core over the other currently identified targets is its remarkable conservation among all six genotypes,  especially in the previously described “homotypic” region of dimerization. Inhibitors optimized on the basis of analogues described here have been found to be equally active on core proteins of genotype 1a or 1b and to inhibit virus production of a HCV 2a strain at nanomolar concentration. Despite several attempts, no resistance mutant were so far found to emerge rapidly in HCV 2a-infected cells grown in the presence of increasing concentrations of core inhibitors. Transketolase is a homodimeric enzyme that catalyses the reversible transfer of two carbons from a ketose donor substrate to an aldose acceptor substrate. Transketolase is the most active enzyme involved into the non-oxidative branch of the pentose phosphate pathway, in charge of generating the ribose molecules necessary for nucleic acid synthesis. Together with the finding that this pathway is highly expressed in the cancer cell, this enzyme provides an excellent target for novel chemotherapeutic agents. Additionally, several crystal structures of this enzyme are available and notably, the human variant of transketolase was recently reported as well allowing the rational structure-based design of human inhibitors. The active centre of transketolase contains a thiamine pyrophosphate cofactor, coordinated to a divalent metal ion, whose binding site has been used for the development of enzyme inhibitors. The most representative inhibitors that mimetize the interactions of thiamine pyrophosphate are oxythiamine and thiamine thiazolone GDC-0199 1257044-40-8 diphosphate. Unfortunately, these compounds lack selectivity as thiamine pyrophosphate is a common cofactor found in multiple enzymes, such as pyruvate dehydrogenase. More recently, several thiamine antagonists were designed with the aim of obtaining more selective inhibitors with Bortezomib improved physical properties. Nonetheless, it is interesting to find additional binding sites allowing drug discovery, not based on the active centre of transketolase but on critical allosteric points of the enzyme. Here, we utilize the homology model of human transketolase recently reported by our group to analyze the hot spot residues of the homodimeric interface and perform a pharmacophore-based virtual screening. This strategy yielded a novel family of compounds, containing the phenyl urea group, as new transketolase inhibitors not based on antagonizing thiamine pyrophosphate. The activity of these compounds, confirmed in transketolase cell extract and in two cancer cell lines, suggests that the phenyl urea scaffold could be used as novel starting point to generate new promising chemotherapeutic agents by targeting human transketolase. During the course of this research, the crystal structure of human transketolase was made public allowing its comparison with our previously reported homology model that was used in the virtual screening protocol. Nonetheless, this sequence is solvent exposed not participating in dimer stabilization nor catalytical activity. It is worth mentioning that the proposed pharmacophore used in this study can be also extracted, with minor distances differences, from the crystal structure of human transketolase.
especially in the previously described “homotypic” region of dimerization. Inhibitors optimized on the basis of analogues described here have been found to be equally active on core proteins of genotype 1a or 1b and to inhibit virus production of a HCV 2a strain at nanomolar concentration. Despite several attempts, no resistance mutant were so far found to emerge rapidly in HCV 2a-infected cells grown in the presence of increasing concentrations of core inhibitors. Transketolase is a homodimeric enzyme that catalyses the reversible transfer of two carbons from a ketose donor substrate to an aldose acceptor substrate. Transketolase is the most active enzyme involved into the non-oxidative branch of the pentose phosphate pathway, in charge of generating the ribose molecules necessary for nucleic acid synthesis. Together with the finding that this pathway is highly expressed in the cancer cell, this enzyme provides an excellent target for novel chemotherapeutic agents. Additionally, several crystal structures of this enzyme are available and notably, the human variant of transketolase was recently reported as well allowing the rational structure-based design of human inhibitors. The active centre of transketolase contains a thiamine pyrophosphate cofactor, coordinated to a divalent metal ion, whose binding site has been used for the development of enzyme inhibitors. The most representative inhibitors that mimetize the interactions of thiamine pyrophosphate are oxythiamine and thiamine thiazolone GDC-0199 1257044-40-8 diphosphate. Unfortunately, these compounds lack selectivity as thiamine pyrophosphate is a common cofactor found in multiple enzymes, such as pyruvate dehydrogenase. More recently, several thiamine antagonists were designed with the aim of obtaining more selective inhibitors with Bortezomib improved physical properties. Nonetheless, it is interesting to find additional binding sites allowing drug discovery, not based on the active centre of transketolase but on critical allosteric points of the enzyme. Here, we utilize the homology model of human transketolase recently reported by our group to analyze the hot spot residues of the homodimeric interface and perform a pharmacophore-based virtual screening. This strategy yielded a novel family of compounds, containing the phenyl urea group, as new transketolase inhibitors not based on antagonizing thiamine pyrophosphate. The activity of these compounds, confirmed in transketolase cell extract and in two cancer cell lines, suggests that the phenyl urea scaffold could be used as novel starting point to generate new promising chemotherapeutic agents by targeting human transketolase. During the course of this research, the crystal structure of human transketolase was made public allowing its comparison with our previously reported homology model that was used in the virtual screening protocol. Nonetheless, this sequence is solvent exposed not participating in dimer stabilization nor catalytical activity. It is worth mentioning that the proposed pharmacophore used in this study can be also extracted, with minor distances differences, from the crystal structure of human transketolase.
The relatively smaller effect of radiation with rapamycin in contrast with the observed synergistic effect of radiation with sunitinib
The antiangiogenic effects were attributed to BAY-60-7550 decreased production of VEGF and resistance of endothelial cells to VEGF stimulation. Further studies used the rapalog RAD001 and compared its effects with known antiangiogenic agents. The results showed that RAD001 was found to be associated with decreasing the tumor vessel density and the maturity of the tumor vessels, whereas the antiangiogenic  drug vatalanib was found to impact only the microvascular density but not the vessel maturity consistent with this class of drugs which impact the VEGF/ VEGFR complex. On the other hand, Zhang et al. reported that the aSMA level, a marker of mature pericytes, increased in Tubulin Acetylation Inducer rapamycin treated tumor compared with non-treated tumor. Other studies have shown that radiation induces activation of mTOR pathways in the tumor endothelial cells making them more sensitive to response with rapamycin. However, a more recent study using a retro-inhibition approach found that HNSCC cells and not the tumor microenvironment as the target for rapamycin activity and that the anti-angiogenic effect is a likely downstream consequence of mTOR inhibition in cancer cells. Imaging of properties intrinsic to tumor physiology such as tumor pO2 and tumor microvessel density made it possible to sequentially follow rapamycin induced changes during the course of treatment non-invasively and sequentially during the treatment course. The key finding in the present study pertaining to the rapamycin effect on tumor physiology is that the tumor microvessel density, when monitored longitudinally showed a significant decrease whereas a transient increase in tumor pO2 was found followed by onset of hypoxia. It should be noted that the USPIO-based blood volume assessment may overestimate the values in tumors because of their leakiness compared to normal tissues. The observations from histological experiments were in agreement with the imaging observations. The rapamycin-induced decrease in CD31 staining was found to be in agreement with imaging experiments where a loss in microvessel density was found. However, there was a small but non-significant decrease in staining of aSMA which reflects the retention of the integrity of the pericyte coverage of the tumor vasculature after rapamycin administration. These results indicate that rapamycin treatment pruned immature blood vessels rather than mature blood vessels. It is expected that these changes in tumor microvasculature can cause improvement of blood flow, a phenomenon known as vascular normalization. The transient increase in the pO2 by rapamycin treatment can be attributed to the increased blood flow in the tumor, which was demonstrated by a 40% increase in tumor initial uptake of Gd-DTPA 2 days after rapamycin treatment in the DCE-MRI study. The identification of transient improvements in tumor oxygenation 2 days after rapamycin treatment provides an opportunity for chemoradiation modalities where radiation therapy can be timed to take advantage of increases in tumor pO2 to elicit improved response. The results in the present study show enhancement in tumor radioresponse by rapamycin treatment. This data suggests that the transiently increased level of median tumor pO2 in rapamycin treated mice compared to the day matched control group may be responsible for the observed effect of radioresponse with combination treatment.
drug vatalanib was found to impact only the microvascular density but not the vessel maturity consistent with this class of drugs which impact the VEGF/ VEGFR complex. On the other hand, Zhang et al. reported that the aSMA level, a marker of mature pericytes, increased in Tubulin Acetylation Inducer rapamycin treated tumor compared with non-treated tumor. Other studies have shown that radiation induces activation of mTOR pathways in the tumor endothelial cells making them more sensitive to response with rapamycin. However, a more recent study using a retro-inhibition approach found that HNSCC cells and not the tumor microenvironment as the target for rapamycin activity and that the anti-angiogenic effect is a likely downstream consequence of mTOR inhibition in cancer cells. Imaging of properties intrinsic to tumor physiology such as tumor pO2 and tumor microvessel density made it possible to sequentially follow rapamycin induced changes during the course of treatment non-invasively and sequentially during the treatment course. The key finding in the present study pertaining to the rapamycin effect on tumor physiology is that the tumor microvessel density, when monitored longitudinally showed a significant decrease whereas a transient increase in tumor pO2 was found followed by onset of hypoxia. It should be noted that the USPIO-based blood volume assessment may overestimate the values in tumors because of their leakiness compared to normal tissues. The observations from histological experiments were in agreement with the imaging observations. The rapamycin-induced decrease in CD31 staining was found to be in agreement with imaging experiments where a loss in microvessel density was found. However, there was a small but non-significant decrease in staining of aSMA which reflects the retention of the integrity of the pericyte coverage of the tumor vasculature after rapamycin administration. These results indicate that rapamycin treatment pruned immature blood vessels rather than mature blood vessels. It is expected that these changes in tumor microvasculature can cause improvement of blood flow, a phenomenon known as vascular normalization. The transient increase in the pO2 by rapamycin treatment can be attributed to the increased blood flow in the tumor, which was demonstrated by a 40% increase in tumor initial uptake of Gd-DTPA 2 days after rapamycin treatment in the DCE-MRI study. The identification of transient improvements in tumor oxygenation 2 days after rapamycin treatment provides an opportunity for chemoradiation modalities where radiation therapy can be timed to take advantage of increases in tumor pO2 to elicit improved response. The results in the present study show enhancement in tumor radioresponse by rapamycin treatment. This data suggests that the transiently increased level of median tumor pO2 in rapamycin treated mice compared to the day matched control group may be responsible for the observed effect of radioresponse with combination treatment.