The work presented here supports core, the HCV capsid protein as a novel target for anti-HCV drug development. We show that an inhibitor of capsid protein dimerization can specifically and directly bind to core and core-based complexes with other HCV proteins. This binding possibly results in disruption of assembly or in disassembly of the viral particle, leading to reduction of infective HCV particles. One added advantage of HCV core over the other currently identified targets is its remarkable conservation among all six genotypes, 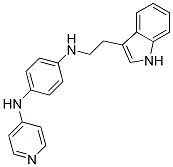 especially in the previously described “homotypic” region of dimerization. Inhibitors optimized on the basis of analogues described here have been found to be equally active on core proteins of genotype 1a or 1b and to inhibit virus production of a HCV 2a strain at nanomolar concentration. Despite several attempts, no resistance mutant were so far found to emerge rapidly in HCV 2a-infected cells grown in the presence of increasing concentrations of core inhibitors. Transketolase is a homodimeric enzyme that catalyses the reversible transfer of two carbons from a ketose donor substrate to an aldose acceptor substrate. Transketolase is the most active enzyme involved into the non-oxidative branch of the pentose phosphate pathway, in charge of generating the ribose molecules necessary for nucleic acid synthesis. Together with the finding that this pathway is highly expressed in the cancer cell, this enzyme provides an excellent target for novel chemotherapeutic agents. Additionally, several crystal structures of this enzyme are available and notably, the human variant of transketolase was recently reported as well allowing the rational structure-based design of human inhibitors. The active centre of transketolase contains a thiamine pyrophosphate cofactor, coordinated to a divalent metal ion, whose binding site has been used for the development of enzyme inhibitors. The most representative inhibitors that mimetize the interactions of thiamine pyrophosphate are oxythiamine and thiamine thiazolone GDC-0199 1257044-40-8 diphosphate. Unfortunately, these compounds lack selectivity as thiamine pyrophosphate is a common cofactor found in multiple enzymes, such as pyruvate dehydrogenase. More recently, several thiamine antagonists were designed with the aim of obtaining more selective inhibitors with Bortezomib improved physical properties. Nonetheless, it is interesting to find additional binding sites allowing drug discovery, not based on the active centre of transketolase but on critical allosteric points of the enzyme. Here, we utilize the homology model of human transketolase recently reported by our group to analyze the hot spot residues of the homodimeric interface and perform a pharmacophore-based virtual screening. This strategy yielded a novel family of compounds, containing the phenyl urea group, as new transketolase inhibitors not based on antagonizing thiamine pyrophosphate. The activity of these compounds, confirmed in transketolase cell extract and in two cancer cell lines, suggests that the phenyl urea scaffold could be used as novel starting point to generate new promising chemotherapeutic agents by targeting human transketolase. During the course of this research, the crystal structure of human transketolase was made public allowing its comparison with our previously reported homology model that was used in the virtual screening protocol. Nonetheless, this sequence is solvent exposed not participating in dimer stabilization nor catalytical activity. It is worth mentioning that the proposed pharmacophore used in this study can be also extracted, with minor distances differences, from the crystal structure of human transketolase.
especially in the previously described “homotypic” region of dimerization. Inhibitors optimized on the basis of analogues described here have been found to be equally active on core proteins of genotype 1a or 1b and to inhibit virus production of a HCV 2a strain at nanomolar concentration. Despite several attempts, no resistance mutant were so far found to emerge rapidly in HCV 2a-infected cells grown in the presence of increasing concentrations of core inhibitors. Transketolase is a homodimeric enzyme that catalyses the reversible transfer of two carbons from a ketose donor substrate to an aldose acceptor substrate. Transketolase is the most active enzyme involved into the non-oxidative branch of the pentose phosphate pathway, in charge of generating the ribose molecules necessary for nucleic acid synthesis. Together with the finding that this pathway is highly expressed in the cancer cell, this enzyme provides an excellent target for novel chemotherapeutic agents. Additionally, several crystal structures of this enzyme are available and notably, the human variant of transketolase was recently reported as well allowing the rational structure-based design of human inhibitors. The active centre of transketolase contains a thiamine pyrophosphate cofactor, coordinated to a divalent metal ion, whose binding site has been used for the development of enzyme inhibitors. The most representative inhibitors that mimetize the interactions of thiamine pyrophosphate are oxythiamine and thiamine thiazolone GDC-0199 1257044-40-8 diphosphate. Unfortunately, these compounds lack selectivity as thiamine pyrophosphate is a common cofactor found in multiple enzymes, such as pyruvate dehydrogenase. More recently, several thiamine antagonists were designed with the aim of obtaining more selective inhibitors with Bortezomib improved physical properties. Nonetheless, it is interesting to find additional binding sites allowing drug discovery, not based on the active centre of transketolase but on critical allosteric points of the enzyme. Here, we utilize the homology model of human transketolase recently reported by our group to analyze the hot spot residues of the homodimeric interface and perform a pharmacophore-based virtual screening. This strategy yielded a novel family of compounds, containing the phenyl urea group, as new transketolase inhibitors not based on antagonizing thiamine pyrophosphate. The activity of these compounds, confirmed in transketolase cell extract and in two cancer cell lines, suggests that the phenyl urea scaffold could be used as novel starting point to generate new promising chemotherapeutic agents by targeting human transketolase. During the course of this research, the crystal structure of human transketolase was made public allowing its comparison with our previously reported homology model that was used in the virtual screening protocol. Nonetheless, this sequence is solvent exposed not participating in dimer stabilization nor catalytical activity. It is worth mentioning that the proposed pharmacophore used in this study can be also extracted, with minor distances differences, from the crystal structure of human transketolase.