It indicated that the aberrant activation of FLT3 and/or drug-resistant FLT3, including pre-existing and acquired drug-resistant mutants, could rarely be fully inhibited by single-agent treatment. Thus, there is a need for the identification of more effective inhibitors of FLT3 and the development of novel therapeutic approaches, including drug combination strategies that target not only FLT3 but also molecules relevant to the FLT3 survival pathway to override current drug resistance. In this study, we demonstrated the efficacy of the novel FLT3 inhibitor BPR1J-340 in various in vitro and in vivo models of AML and identify synergistic effects with HDACi SAHA on the cytotoxicity of FL3-ITD-expressing cells in in vitro analyses. Previously, we identified a sulfonamide series of 3-phenyl-1H-5pyrazolylamine-based compounds as potent inhibitors of FLT3 such as BPR1J-097. In continuing to our efforts to develop potent FLT3 inhibitors, we intended to search other series of inhibitors that not only increased the in vitro growth-inhibitory effect on AML cells but also prolonged the duration of action in vivo. Through rational design, we discovered BPR1J-340, which is a urea series of 3-phenyl-1H-5-pyrazolylamine-based FLT3 inhibitor, with effectively inhibits FLT3-WT or FLT3-ITD activity in vitro and in vivo. Because multiple signaling pathways influence the growth and metastatic potential of tumor cells, many of the inhibitors in clinical development are designed as multi-targeted inhibitors that block a limited number of oncogenic kinases. Thus, the kinase selectivity Nilotinib profiling of BPR1J-340 was performed to identify additional targets in a panel of 59 tested oncogenic kinases. In further biochemical assay, BPR1J-340 demonstrated potent inhibitionagainst the angiogenic kinases VEGFR1, VEGFR2, and VEGFR3, which all play an important role in the tumor microenvironment. In addition, BPR1J-340 potently inhibited TRKA activity with an IC50 value of 8 nM. Taken together, BPRJ-340 is characterized as a selective multi-targeted inhibitor with potent inhibition activity against FLT3-WT, FLT3-D835Y, VEGFR2, VEGFR3, and TRKA. This inhibition profile may allow BPRJ-340 to inhibit tumor growth directly by blocking the aberrant FLT3 signaling pathway and indirectly by targeting tumor angiogenesis. BPR1J-340 may also have clinical potential in tumor driven by abnormally expressed TRKA receptors, which can occur in brain, prostate, pancreatic, and breast cancer. BPR-1J340 inhibited cellular FLT3 PI-103 PI3K inhibitor phosphorylation and modulated the FLT3 signaling pathway, which resulted in inhibition of proliferation and induction of apoptosis. BPR1J-340 demonstrated potent growth inhibition, predominantly in FLT3dependent cellsbut not in FLT3independent cells. BPR1J-340 inhibited the proliferation of mutant FLT3-ITD + cells with an GC50 of 2�C6 nM and inhibited FLT3ITD phosphorylation with an IC50 of 10 nM; even the cells transfected with the FLT3-D835Y mutant were also inhibited by BPR1J-340 with an IC50 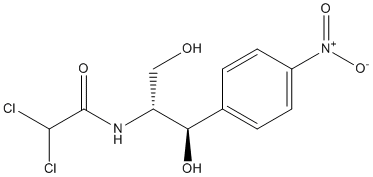 of approximately 100 nM. Consistent with these results, BPR1J-340 effectively induced apoptosis in FLT3-ITD + cells. HDAC inhibitors may exhibit growth inhibition activity against AML cells and significantly improve the therapeutic efficacy of FLT3 inhibitors. A recent study reported that HDACi LBH589 plus an FLT3 inhibitorcombination treatment could synergistically induce apoptosis through FLT3ITD and STAT5 degradation. It also demonstrated that activated caspase-3contributes to the degrdation of FLT3-ITD and STAT5. FLT3-ITD degradation was also reported secondary to HDACi-induced up-regulation of UBCH8 and down-regulation of HSP90.
of approximately 100 nM. Consistent with these results, BPR1J-340 effectively induced apoptosis in FLT3-ITD + cells. HDAC inhibitors may exhibit growth inhibition activity against AML cells and significantly improve the therapeutic efficacy of FLT3 inhibitors. A recent study reported that HDACi LBH589 plus an FLT3 inhibitorcombination treatment could synergistically induce apoptosis through FLT3ITD and STAT5 degradation. It also demonstrated that activated caspase-3contributes to the degrdation of FLT3-ITD and STAT5. FLT3-ITD degradation was also reported secondary to HDACi-induced up-regulation of UBCH8 and down-regulation of HSP90.
Month: July 2019
Dual inhibition of Etk and Src could overcome this disadvantage and induce massive
Inhibition of Src leads to cell cycle arrest and reduction of migration and inhibition. Dual inhibition of Src and Etk are expected to inhibit growth, migration, and survival of the cells. To the best of our knowledge, this is the first selective Etk/Src inhibitor reported to date. CTA095 belongs to a novel class of kinase inhibitors with a chemical structure distinct from other known TKIs. This inhibitor is most potent in inhibiting Etk followed by Src, but it is not a potent inhibitor for other Tec and Src family kinases such as Btk, Yes and Lyn. Treatment of PC3 cells with CTA095 resulted in the decrease of phoshorylation of Etk and Src, as well as the downstream signals Stat3 and Akt. CTA095 has little effect on the survival of the immortalized normal prostate cell RWPE1, suggesting that the growth and survival of prostate cancer cells are more dependent on Etk and Src signals, consistent with the observed overexpression of Etk in prostate cancer tissues and the development of PIN phenotype in transgenic animals carrying overexpressed Etk. Etk interacts with and inactivates p53, and overexpression of Etk in prostate cancer cells confers resistance to androgen deprivationand photodynamic therapy. On the other hand, truncated Etk, due to caspase cleavage, induces apoptosis. This data taken together suggests that Etk is a strong regulator of apoptosis. At the same time, CTA095 was found to induce autophagy in human prostate cancer cells. Autophagy is generally regarded as a pro-survival mechanism, where wasted proteins and retired organs are degraded to regenerate energy during stress conditions. It is inhibited by energy sensor  mTOR, a target of rapamycin. Excessive autophagy, under special conditions, however, can lead to programmed cell death. Previously, we reported that autophagy induced by Src inhibitor is the underlying cause of apoptosis-resistance of the GW786034 treated cells. These cells are growth arrested, but do not undergo apoptosis. Blocking autophagy by siRNA BKM120 targeting of autophagy components, or the simultaneous application of CQ, an inhibitor of autophagy flow, results in massive cell death. It is interesting that, like the Src inhibitor, CTA095 induced autophagy is likely due to its inhibition of mTOR activation. However, unlike the Src inhibitor, CTA095 inhibits both Etk and Src, and can overcome the apoptosis resistance induced by autophagy. This data suggests that the absence of Etk proactively turns on the apoptosis pathway, which cannot be completely overridden by the restoring act of autophagy. The finding that the autophagy inhibitor CQ further enhances the apoptosis-inducing effect of CTA095 suggests that autophagy contributes partially to the survival of CTA095 treated cells. Alternatively, CQ is known to have other cellular effects including induction of p53 and intercalation of DNA, which may contribute additional toxicity to the treated cells. Curiously, we found that the autophagy inducer RPM also synergizes with CTA095, suggesting excessive autophagy may contribute additional means to kill prostate cancer cells. CTA095 was also a chemo-sensitizer and showed synergistic effect with PTX, indicating its role in combination therapy for prostate cancer. In addition to these findings, CTA095 was found to inhibit HUVEC cell tube formation and “wound healing” of prostate cancer cells, implying its role in inhibition of angiogenesis and metastasis of human prostate cancer cells. More interestingly, simultaneously targeting of Etk and Src by CTA095 overcomes the Src inhibitor resistance in prostate cancer cells. The Src inhibitor AZD0530 has a good effect on inhibition of bone metastasis of prostate cancer, but lacks the ability to induce apoptosis in prostate cancer cells.
mTOR, a target of rapamycin. Excessive autophagy, under special conditions, however, can lead to programmed cell death. Previously, we reported that autophagy induced by Src inhibitor is the underlying cause of apoptosis-resistance of the GW786034 treated cells. These cells are growth arrested, but do not undergo apoptosis. Blocking autophagy by siRNA BKM120 targeting of autophagy components, or the simultaneous application of CQ, an inhibitor of autophagy flow, results in massive cell death. It is interesting that, like the Src inhibitor, CTA095 induced autophagy is likely due to its inhibition of mTOR activation. However, unlike the Src inhibitor, CTA095 inhibits both Etk and Src, and can overcome the apoptosis resistance induced by autophagy. This data suggests that the absence of Etk proactively turns on the apoptosis pathway, which cannot be completely overridden by the restoring act of autophagy. The finding that the autophagy inhibitor CQ further enhances the apoptosis-inducing effect of CTA095 suggests that autophagy contributes partially to the survival of CTA095 treated cells. Alternatively, CQ is known to have other cellular effects including induction of p53 and intercalation of DNA, which may contribute additional toxicity to the treated cells. Curiously, we found that the autophagy inducer RPM also synergizes with CTA095, suggesting excessive autophagy may contribute additional means to kill prostate cancer cells. CTA095 was also a chemo-sensitizer and showed synergistic effect with PTX, indicating its role in combination therapy for prostate cancer. In addition to these findings, CTA095 was found to inhibit HUVEC cell tube formation and “wound healing” of prostate cancer cells, implying its role in inhibition of angiogenesis and metastasis of human prostate cancer cells. More interestingly, simultaneously targeting of Etk and Src by CTA095 overcomes the Src inhibitor resistance in prostate cancer cells. The Src inhibitor AZD0530 has a good effect on inhibition of bone metastasis of prostate cancer, but lacks the ability to induce apoptosis in prostate cancer cells.
Hydrogen bonds with the amide group of Thr36 in the uracil binding pocket and have interactions with the Asp35�CArg37 salt bridge
Additional stabilization is achieved through stable cation-p interactions between their phenyl rings and the positively charged guanidino group of Arg37. The same interactions are also observed in the cocrystal structure of compound 1b. The introduction of substituted benzoic acid derivatives as glutamic acid mimetics in the second-generation sulfonamide inhibitors allows p�Cp stacking interactions with the Phe422 phenyl ring. This might contribute to increased binding affinities compared to the D-Glu-containing compounds. Another important difference between the binding modes of the most potent compound from the first-generation 1b and of the most potent compound from the second-generation 6b that could contribute to the 10-fold difference in their inhibitory activities lies in interactions with the central domain residues. Only indirect interactions of the ligand sulfonyl group across the water molecule with the residues Asn138 and Ser159 are observed in the crystal structure of the 1b�CMurD complex. The MD simulations show that the direct hydrogen bond of compound 1b with Asn138 is formed much less frequently compared to the case of compound 6b. These observations are supported by NMR data. The CSPs�� patterns reveal a significantly increased effect of compound 6b on the central domain signals with regard to the compound 1b. The MD data reveal complex dynamic behaviors of these ligand�CMurD complexes and show that these influence the ligand�C enzyme contacts. As well as the rotation of ligand segments at the MurD binding site, as revealed by transferred NOESY, slight opening/closing movements of the protein domains are seen in MD trajectories. Movements of protein domains can adversely affect ligand binding through effects on the conformation and flexibility of the bound ligand, the stability of the ligand�Cenzyme interactions, and the binding-site adaptability. These movements should not be confused with the open and closed conformations of the MurD protein that have  been reported in the literature, where the Cterminal domain has a drastically different position. The most pronounced fluctuations are evident from the distances between the geometric centers of the C-terminal and N-terminal domains. The Regorafenib variations between the minimal and maximal distances can be up to 5.5 A ?. An additional, longerMD run of selected ligands 6a and 6b clearly indicate that the fluctuations of distance between these geometric centers are less pronounced when the most potent inhibitor 6b is bound to MurD. This might be the consequence of better binding interactions that tend to hold the domains together. Visual inspection of the trajectories reveals that the movements of the C-terminal and N-terminal domains have important roles in ligand binding. Sulfonamide inhibitors span from the N-terminal domain to the C-terminal domain. Opening movements tend to weaken the interactions either with the uracil binding pocket or with the D-Glu binding site. The C6-alkyloxy-substituted compoundsgenerally have weaker interactions with the uracil binding pocket compared to the C6-arylalkyloxy-substituted compounds. Consequently, the alkyl WZ8040 EGFR/HER2 inhibitor chains can even be pulled out of the uracil binding pocket during domain movement. The C6-alkyloxy-substituted compounds are also shorter than the C6-arylalkyloxy-substituted compounds. Therefore, the alkyl chain has more freedom to move in the uracil binding pocket. This is accompanied by conformational changes of the ligand. During domain movement, a rotation of the mimetic ring for compounds 3a, 4a, and 5a is observed around the hinges formed by the carboxyl groups that switches their conformation between extended and bent form.
been reported in the literature, where the Cterminal domain has a drastically different position. The most pronounced fluctuations are evident from the distances between the geometric centers of the C-terminal and N-terminal domains. The Regorafenib variations between the minimal and maximal distances can be up to 5.5 A ?. An additional, longerMD run of selected ligands 6a and 6b clearly indicate that the fluctuations of distance between these geometric centers are less pronounced when the most potent inhibitor 6b is bound to MurD. This might be the consequence of better binding interactions that tend to hold the domains together. Visual inspection of the trajectories reveals that the movements of the C-terminal and N-terminal domains have important roles in ligand binding. Sulfonamide inhibitors span from the N-terminal domain to the C-terminal domain. Opening movements tend to weaken the interactions either with the uracil binding pocket or with the D-Glu binding site. The C6-alkyloxy-substituted compoundsgenerally have weaker interactions with the uracil binding pocket compared to the C6-arylalkyloxy-substituted compounds. Consequently, the alkyl WZ8040 EGFR/HER2 inhibitor chains can even be pulled out of the uracil binding pocket during domain movement. The C6-alkyloxy-substituted compounds are also shorter than the C6-arylalkyloxy-substituted compounds. Therefore, the alkyl chain has more freedom to move in the uracil binding pocket. This is accompanied by conformational changes of the ligand. During domain movement, a rotation of the mimetic ring for compounds 3a, 4a, and 5a is observed around the hinges formed by the carboxyl groups that switches their conformation between extended and bent form.
Transient induction fulfilled the requirement of being pharmacodynamic biomarkers for this radiosensitizing drug in fractionated radiotherapy
Among the identified candidate genes, MYC repression was found in all patient samples and tested experimental conditions, possibly underscoring the impact of the myc protooncogene in this particular therapeutic setting. Histones are small proteins critical to the efficient packaging of DNA in the nucleus. DNA winds itself around the surface of the histones, forming DNA-protein complexes known as nucleosomes. The N-terminal histone tail protrudes from the nucleosome, allowing for post-translational modification of key histone residues. These post-translational modifications commonly consist of phosphorylation, acetylation, methylation, ubiquitylation, ribosylation, and sumoylation, to name a few. Combinations of such histone modifications take part in the regulation of DNA transcription and constitute an additional level to the genetic code, termed the “histone code”. These modifications are dynamically maintained by various histone-modifying enzymes that control their transfer and removal. While histone-modifying enzymes are important for normal cell function, overexpression of the enzymes can result in the aberrant silencing of genes that are required to govern cell identity. For example, enhancer of zeste homolog 2 is a SET-domain containing histone methyltransferase that catalyzes the diand trimethylation of the lysine at position 27 of histone H3. Both methylation states of H3K27 have been linked to heterochromatic genomic regions and to epigenetic silencing. Overabundance of EZH2 has been linked to the silencing of more than 100 genes in prostate  cell lines, including several important tumor suppressors. For this reason, the overexpression of EZH2 has been correlated to the invasiveness of breast and prostate cancer and linked to various other cancer types. Moreover, recurrent mutations of EZH2 have been found in germinal center B-cell like diffuse large B-cell lymphoma, follicular lymphoma, and melanoma. The mutated residues alter the substrate specificity of EZH2 and facilitate the conversion from a dimethylated to a trimethylated state, thus resulting in significantly elevated global H3K27me3 levels. Cancer cells harboring EZH2 mutations were recently shown to be dependent on the EZH2 catalytic activity since their viability was severely affected by EZH2 small molecule inhibitors. Additionally, studies have shown that RNAi mediated knockdown of EZH2 inhibits the growth and migration of cancer cells and upregulates the tumor suppressor gene BRCA1. This makes the inhibition of histone-modifying enzymes, in particular EZH2, an important target in the development of cancer therapeutics for many different cancer types. Histone methyltransferase small molecule inhibitors obtained through random, large-scale screening of compound libraries have been reported in the literature. However, the number of potent and selective inhibitors remains small and the community still does not have Doxorubicin adequate tools to target all methyltransferases that are implicated in human disease. For this reason EZH2 remains an important target for inhibitor design. The pharmacological properties of peptidic inhibitors make their use in the development of cancer therapeutics TWS119 difficult. However, the specificity with which they can act with their binding partner make them desirable for the development of chemical probes for the interrogation of methyltransferase and chromatin biology. Peptide inhibitors are generally more specific than small molecule inhibitors as they often more closely resemble the natural binders of many target proteins. The aim of this work was to find specific peptidic inhibitors of EZH2 using a computational de novo peptide design framework.
cell lines, including several important tumor suppressors. For this reason, the overexpression of EZH2 has been correlated to the invasiveness of breast and prostate cancer and linked to various other cancer types. Moreover, recurrent mutations of EZH2 have been found in germinal center B-cell like diffuse large B-cell lymphoma, follicular lymphoma, and melanoma. The mutated residues alter the substrate specificity of EZH2 and facilitate the conversion from a dimethylated to a trimethylated state, thus resulting in significantly elevated global H3K27me3 levels. Cancer cells harboring EZH2 mutations were recently shown to be dependent on the EZH2 catalytic activity since their viability was severely affected by EZH2 small molecule inhibitors. Additionally, studies have shown that RNAi mediated knockdown of EZH2 inhibits the growth and migration of cancer cells and upregulates the tumor suppressor gene BRCA1. This makes the inhibition of histone-modifying enzymes, in particular EZH2, an important target in the development of cancer therapeutics for many different cancer types. Histone methyltransferase small molecule inhibitors obtained through random, large-scale screening of compound libraries have been reported in the literature. However, the number of potent and selective inhibitors remains small and the community still does not have Doxorubicin adequate tools to target all methyltransferases that are implicated in human disease. For this reason EZH2 remains an important target for inhibitor design. The pharmacological properties of peptidic inhibitors make their use in the development of cancer therapeutics TWS119 difficult. However, the specificity with which they can act with their binding partner make them desirable for the development of chemical probes for the interrogation of methyltransferase and chromatin biology. Peptide inhibitors are generally more specific than small molecule inhibitors as they often more closely resemble the natural binders of many target proteins. The aim of this work was to find specific peptidic inhibitors of EZH2 using a computational de novo peptide design framework.
Inhibition was enzyme specific as iGP-1 had no effect on NAD-linked cGPDH activity
This is not surprising considering the two forms are distinct in all respects except their ability to oxidize glycerol 3-phosphate to DHAP. However, this enzyme specificity suggests that iGPs are likely not acting as inhibitory analogs in the substrate binding pocket as described for non-selective inhibitors of both forms such as glyceraldehyde 3-phosphate. The MDV3100 observation of increased Km as well as the lower values for Kic than Kiu indicates that both iGP-1 and iGP-5 have a greater affinity for free enzyme. Both inhibitors have Hill slopes near unity, suggesting they interact with mGPDH at a single, allosteric binding site. Although the analysis of inhibition kinetics was performed in the presence of activating calcium, our evidence from assays of mGPDH-specific H2O2 production and DYm driven by glycerol phosphate suggest that iGPs act independently of the calcium-sensing mechanism of mGPDH. We cannot rule out direct interactions between iGPs and either the FAD binding domain in the soluble portion of mGPDH or the ubiquinone binding  pocket embedded in the outer leaflet of the inner mitochondrial membrane. The interaction with the ubiquinone pocket might be tested by studies similar to those presented here but with differing ubiquinol as the electron donor to the enzyme. Future co-crystallization structural studies and enzymatic assays of iGPs with the bacterial or mammalian FAD-linked GPDH may provide the best opportunities to identify the exact mode of interaction and mechanism of action of these novel inhibitors. As initial confirmed hits in a small-molecule screen, our most promising iGPs demonstrate excellent potency and good selectivity. Apart from a subtle effect on succinate oxidation at high concentrations, iGP-1 does not alter mitochondrial oxidation of numerous substrates including a second dicarboxylate, malate. Therefore, it is unlikely that the subtle effect on succinate oxidation is due to inhibition of the dicarboxylate transporter by the succinamide of iGPs. Indeed, analogs iGP-17�C iGP-19 retain the succinamide without or with the attached phenyl ring yet do not alter succinate oxidation as assessed by H2O2 production by succinate alone. This suggests at least partial dependence on the benzimidazole ring system for the subtle effect of iGP-1 on succinate oxidation, perhaps via direct interaction with complex II. Reactions of the tricarboxylic acid cycle shared between succinate, malate, and pyruvate oxidation also do not appear to be affected by iGP-1. Further, iGP-1 shows no effects on the maintenance of proton motive force or rates of ATP synthesis with substrates other than glycerol phosphate. In addition, we can infer from the synaptosomal experiments that iGP-1 does not prevent pyruvate uptake into cells or mitochondria and does not directly alter glycolysis. Therefore, our data identify an exemplary inhibitor that is both potent and selective against mGPDH and offers structural targets through which additional improvements to these activities can be BKM120 structure achieved. In conclusion, we have identified a novel class of potent, selective, cell-permeant inhibitors of mGPDH that act via mixed inhibition. Further tests of the role of mGPDH and glycerol phosphate shuttle activities under conditions of neuronal activity or in other cell types with differing shuttle capacities will help determine those in which mGPDH activity is essential. Our novel inhibitors of mGPDH provide means to test these possibilities pharmacologically. The ubiquitin�Cproteasome system is the major protein degradation pathway in every cell. The eukaryotic proteasome is a potential target for antitumor drugs. To date, two proteasome inhibitors, the Bortezomib and Carfilzomib, have been approved for the treatment of multiple myeloma.
pocket embedded in the outer leaflet of the inner mitochondrial membrane. The interaction with the ubiquinone pocket might be tested by studies similar to those presented here but with differing ubiquinol as the electron donor to the enzyme. Future co-crystallization structural studies and enzymatic assays of iGPs with the bacterial or mammalian FAD-linked GPDH may provide the best opportunities to identify the exact mode of interaction and mechanism of action of these novel inhibitors. As initial confirmed hits in a small-molecule screen, our most promising iGPs demonstrate excellent potency and good selectivity. Apart from a subtle effect on succinate oxidation at high concentrations, iGP-1 does not alter mitochondrial oxidation of numerous substrates including a second dicarboxylate, malate. Therefore, it is unlikely that the subtle effect on succinate oxidation is due to inhibition of the dicarboxylate transporter by the succinamide of iGPs. Indeed, analogs iGP-17�C iGP-19 retain the succinamide without or with the attached phenyl ring yet do not alter succinate oxidation as assessed by H2O2 production by succinate alone. This suggests at least partial dependence on the benzimidazole ring system for the subtle effect of iGP-1 on succinate oxidation, perhaps via direct interaction with complex II. Reactions of the tricarboxylic acid cycle shared between succinate, malate, and pyruvate oxidation also do not appear to be affected by iGP-1. Further, iGP-1 shows no effects on the maintenance of proton motive force or rates of ATP synthesis with substrates other than glycerol phosphate. In addition, we can infer from the synaptosomal experiments that iGP-1 does not prevent pyruvate uptake into cells or mitochondria and does not directly alter glycolysis. Therefore, our data identify an exemplary inhibitor that is both potent and selective against mGPDH and offers structural targets through which additional improvements to these activities can be BKM120 structure achieved. In conclusion, we have identified a novel class of potent, selective, cell-permeant inhibitors of mGPDH that act via mixed inhibition. Further tests of the role of mGPDH and glycerol phosphate shuttle activities under conditions of neuronal activity or in other cell types with differing shuttle capacities will help determine those in which mGPDH activity is essential. Our novel inhibitors of mGPDH provide means to test these possibilities pharmacologically. The ubiquitin�Cproteasome system is the major protein degradation pathway in every cell. The eukaryotic proteasome is a potential target for antitumor drugs. To date, two proteasome inhibitors, the Bortezomib and Carfilzomib, have been approved for the treatment of multiple myeloma.