It indicated that the aberrant activation of FLT3 and/or drug-resistant FLT3, including pre-existing and acquired drug-resistant mutants, could rarely be fully inhibited by single-agent treatment. Thus, there is a need for the identification of more effective inhibitors of FLT3 and the development of novel therapeutic approaches, including drug combination strategies that target not only FLT3 but also molecules relevant to the FLT3 survival pathway to override current drug resistance. In this study, we demonstrated the efficacy of the novel FLT3 inhibitor BPR1J-340 in various in vitro and in vivo models of AML and identify synergistic effects with HDACi SAHA on the cytotoxicity of FL3-ITD-expressing cells in in vitro analyses. Previously, we identified a sulfonamide series of 3-phenyl-1H-5pyrazolylamine-based compounds as potent inhibitors of FLT3 such as BPR1J-097. In continuing to our efforts to develop potent FLT3 inhibitors, we intended to search other series of inhibitors that not only increased the in vitro growth-inhibitory effect on AML cells but also prolonged the duration of action in vivo. Through rational design, we discovered BPR1J-340, which is a urea series of 3-phenyl-1H-5-pyrazolylamine-based FLT3 inhibitor, with effectively inhibits FLT3-WT or FLT3-ITD activity in vitro and in vivo. Because multiple signaling pathways influence the growth and metastatic potential of tumor cells, many of the inhibitors in clinical development are designed as multi-targeted inhibitors that block a limited number of oncogenic kinases. Thus, the kinase selectivity Nilotinib profiling of BPR1J-340 was performed to identify additional targets in a panel of 59 tested oncogenic kinases. In further biochemical assay, BPR1J-340 demonstrated potent inhibitionagainst the angiogenic kinases VEGFR1, VEGFR2, and VEGFR3, which all play an important role in the tumor microenvironment. In addition, BPR1J-340 potently inhibited TRKA activity with an IC50 value of 8 nM. Taken together, BPRJ-340 is characterized as a selective multi-targeted inhibitor with potent inhibition activity against FLT3-WT, FLT3-D835Y, VEGFR2, VEGFR3, and TRKA. This inhibition profile may allow BPRJ-340 to inhibit tumor growth directly by blocking the aberrant FLT3 signaling pathway and indirectly by targeting tumor angiogenesis. BPR1J-340 may also have clinical potential in tumor driven by abnormally expressed TRKA receptors, which can occur in brain, prostate, pancreatic, and breast cancer. BPR-1J340 inhibited cellular FLT3 PI-103 PI3K inhibitor phosphorylation and modulated the FLT3 signaling pathway, which resulted in inhibition of proliferation and induction of apoptosis. BPR1J-340 demonstrated potent growth inhibition, predominantly in FLT3dependent cellsbut not in FLT3independent cells. BPR1J-340 inhibited the proliferation of mutant FLT3-ITD + cells with an GC50 of 2�C6 nM and inhibited FLT3ITD phosphorylation with an IC50 of 10 nM; even the cells transfected with the FLT3-D835Y mutant were also inhibited by BPR1J-340 with an IC50 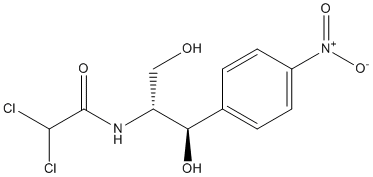 of approximately 100 nM. Consistent with these results, BPR1J-340 effectively induced apoptosis in FLT3-ITD + cells. HDAC inhibitors may exhibit growth inhibition activity against AML cells and significantly improve the therapeutic efficacy of FLT3 inhibitors. A recent study reported that HDACi LBH589 plus an FLT3 inhibitorcombination treatment could synergistically induce apoptosis through FLT3ITD and STAT5 degradation. It also demonstrated that activated caspase-3contributes to the degrdation of FLT3-ITD and STAT5. FLT3-ITD degradation was also reported secondary to HDACi-induced up-regulation of UBCH8 and down-regulation of HSP90.
of approximately 100 nM. Consistent with these results, BPR1J-340 effectively induced apoptosis in FLT3-ITD + cells. HDAC inhibitors may exhibit growth inhibition activity against AML cells and significantly improve the therapeutic efficacy of FLT3 inhibitors. A recent study reported that HDACi LBH589 plus an FLT3 inhibitorcombination treatment could synergistically induce apoptosis through FLT3ITD and STAT5 degradation. It also demonstrated that activated caspase-3contributes to the degrdation of FLT3-ITD and STAT5. FLT3-ITD degradation was also reported secondary to HDACi-induced up-regulation of UBCH8 and down-regulation of HSP90.